Note
Go to the end to download the full example code.
Geometry optimization (LBFGS)¶
Two-stage geometry optimization of a slightly perturbed silicon unit cell.
First, positions are relaxed at fixed cell with
ase.optimize.LBFGS. Then the cell itself is relaxed jointly
with the atomic positions by wrapping the Atoms in
ase.filters.FrechetCellFilter.
The script records the maximum force and total energy at every step and plots them so that the convergence of the two stages is visible at a glance.
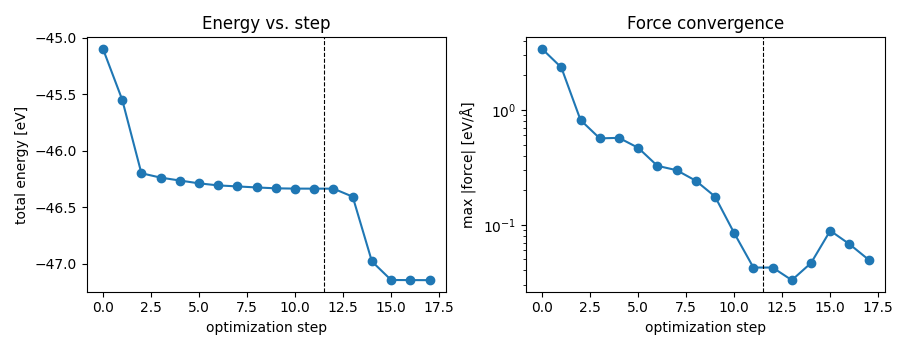
Step Time Energy fmax
LBFGS: 0 16:04:29 -45.099880 3.402630
LBFGS: 1 16:04:29 -45.548882 2.344578
LBFGS: 2 16:04:29 -46.199654 0.816876
LBFGS: 3 16:04:29 -46.238434 0.565600
LBFGS: 4 16:04:29 -46.264095 0.571423
LBFGS: 5 16:04:29 -46.289150 0.468046
LBFGS: 6 16:04:29 -46.307289 0.326138
LBFGS: 7 16:04:29 -46.316319 0.298761
LBFGS: 8 16:04:29 -46.325409 0.241908
LBFGS: 9 16:04:29 -46.333160 0.176208
LBFGS: 10 16:04:29 -46.335651 0.084599
LBFGS: 11 16:04:29 -46.336029 0.042396
Step Time Energy fmax
LBFGS: 0 16:04:29 -46.336029 1.307608
LBFGS: 1 16:04:29 -46.407188 1.285505
LBFGS: 2 16:04:30 -46.979885 0.576498
LBFGS: 3 16:04:30 -47.144043 0.084717
LBFGS: 4 16:04:30 -47.144936 0.064519
LBFGS: 5 16:04:30 -47.145432 0.047221
import matplotlib.pyplot as plt
import numpy as np
from ase.build import bulk
from ase.filters import FrechetCellFilter
from ase.optimize import LBFGS
from upet.calculator import UPETCalculator
atoms = bulk("Si", cubic=True, a=5.43, crystalstructure="diamond")
# perturb positions and cell so the optimizer has something to do
atoms.rattle(0.1, seed=0) # ASE's built-in random displacement method
atoms.set_cell(atoms.cell * 1.05, scale_atoms=True)
calculator = UPETCalculator(model="pet-mad-xs", version="1.5.0", device="cpu")
atoms.calc = calculator
history = {"stage": [], "energy": [], "fmax": []} # type: ignore
def record(stage_name):
def _cb():
results = calculator.results
history["stage"].append(stage_name)
history["energy"].append(float(results["energy"]))
history["fmax"].append(float(np.linalg.norm(results["forces"], axis=1).max()))
return _cb
# stage 1: positions only
opt_pos = LBFGS(atoms)
opt_pos.attach(record("positions"), interval=1)
opt_pos.run(fmax=0.05, steps=30)
# stage 2: joint position + cell relaxation
filtered = FrechetCellFilter(atoms)
opt_cell = LBFGS(filtered)
opt_cell.attach(record("cell"), interval=1)
opt_cell.run(fmax=0.05, steps=30)
steps = np.arange(len(history["energy"]))
stages = np.array(history["stage"])
boundary = (
int(np.searchsorted(stages == "cell", True))
if (stages == "cell").any()
else len(stages)
)
fig, (ax_e, ax_f) = plt.subplots(1, 2, figsize=(9, 3.5))
ax_e.plot(steps, history["energy"], "o-")
ax_e.axvline(boundary - 0.5, color="k", ls="--", lw=0.8)
ax_e.set_xlabel("optimization step")
ax_e.set_ylabel("total energy [eV]")
ax_e.set_title("Energy vs. step")
ax_f.semilogy(steps, history["fmax"], "o-")
ax_f.axvline(boundary - 0.5, color="k", ls="--", lw=0.8)
ax_f.set_xlabel("optimization step")
ax_f.set_ylabel("max |force| [eV/Å]")
ax_f.set_title("Force convergence")
fig.tight_layout()
plt.show()