Note
Go to the end to download the full example code.
Computing LODE descriptors¶
- Authors:
Michele Ceriotti @ceriottm
This notebook demonstrates the use of some advanced features of
torch-pme to compute long-distance equivariants (LODE) features
as in Grisafi and Ceriotti, J. Chem. Phys. (2019)
and Huguenin-Dumittan et al., J. Phys. Chem. Lett. (2023).
Note that a compiled-language CPU implementation of LODE features is
also available in the featomic package.
import ase
import chemiscope
import matplotlib
import numpy as np
import scipy
import torch
from matplotlib import pyplot as plt
import torchpme
from torchpme.potentials import CoulombPotential, Potential, SplinePotential
device = "cpu"
dtype = torch.float64
rng = torch.Generator()
rng.manual_seed(42)
<torch._C.Generator object at 0x7f0082d1c170>
Long-distance equivariant descriptors¶
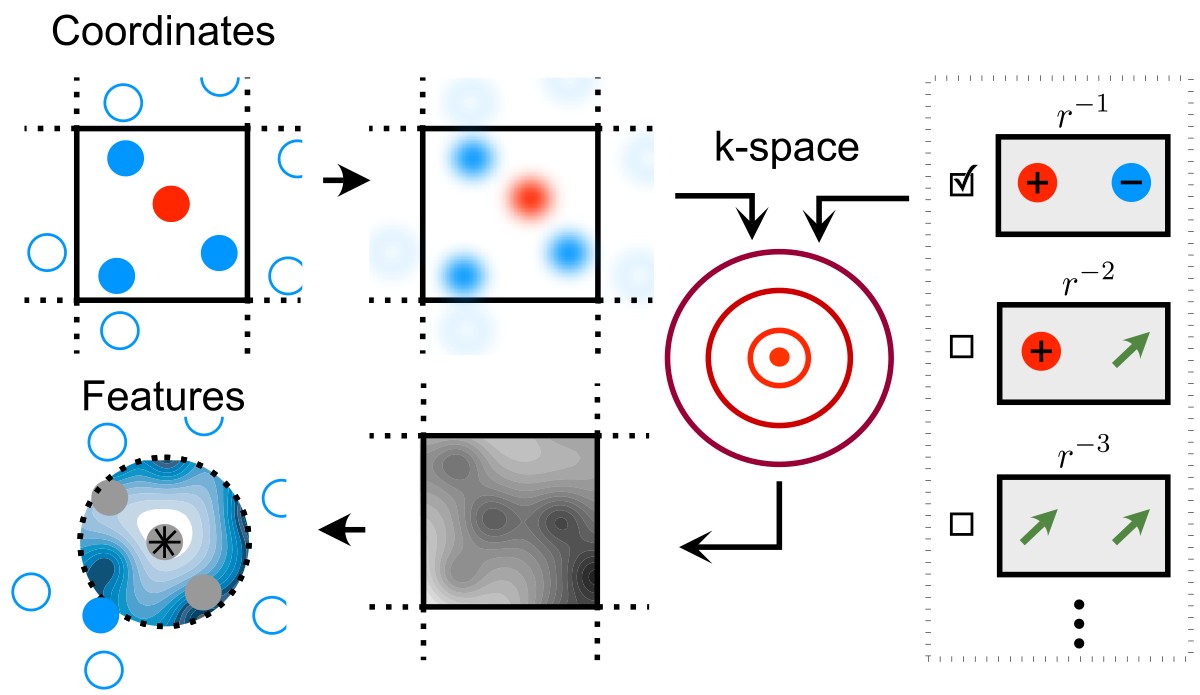
A schematic view of the process of evaluating LODE features. Rather than computing an expansion of the neighbor density (the operation that underlies short-range models, from SOAP to NICE) one first transforms the density in the Fourier domain, then back to obtain a real-space “potential field” that is then expanded on an atom-centered basis.¶
The basic idea behind the LODE framework is to evaluate a “potential field”, convoluting the neighbor density with a suitable kernel
and then expand it on an atom-centered basis, so as to obtain a set of features that describe the environment of each atom.
By choosing a slowly-decaying kernel that emphasizes long-range correlations, and that and that is consistent with the asymptotic behavior of e.g. electrostatic interactions, one achieves a description of the long-range interaction, rather than of the immediate vicinity of each atom. By choosing a basis of spherical harmonics for the angular part, one achieves descriptors that transform as irreducible representations of the rotation group.
Initialize a trial structure¶
We use as an example a distorted rocksalt structure with perturbed positions and charges
structure = ase.Atoms(
positions=[
[0, 0, 0],
[3, 0, 0],
[0, 3, 0],
[3, 3, 0],
[0, 0, 3],
[3, 0, 3],
[0, 3, 3],
[3, 3, 3],
],
cell=[6, 6, 6],
symbols="NaClClNaClNaNaCl",
)
structure = structure.repeat([3, 3, 3])
displacement = torch.normal(
mean=0.0, std=2.5e-1, size=(len(structure), 3), generator=rng
)
structure.positions += displacement.numpy()
charges = torch.tensor(
[[1.0], [-1.0], [-1.0], [1.0], [-1.0], [1.0], [1.0], [-1.0]]
* (len(structure) // 8),
dtype=dtype,
device=device,
).reshape(-1, 1)
charges += torch.normal(mean=0.0, std=1e-1, size=(len(charges), 1), generator=rng)
positions = torch.from_numpy(structure.positions).to(device=device, dtype=dtype)
cell = torch.from_numpy(structure.cell.array).to(device=device, dtype=dtype)
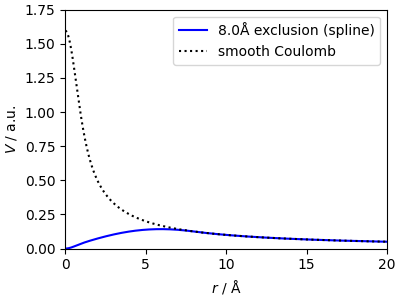
An “excluded-range” smooth Coulomb potential¶
We use SplinePotential to
compute a smooth Coulomb potential with the “short-range” part cut out.
This is important as otherwise the potential carries information on the
local atomic arrangement, which is redundant (as it is usually described
by another part of the model).
CoulombPotential does this by
first computing the potential generated by Gaussian densities, and then
removing in real space the contributions in the vicinity of each atom.
For LODE we must get the potential directly on the grid, and so it is
better to use a numerical kernel that achieves this using only k-space
operations.
smearing = 0.5
exclusion_radius = 8.0
coulomb = CoulombPotential(smearing=smearing, exclusion_radius=None)
coulomb_exclude = CoulombPotential(smearing=smearing, exclusion_radius=exclusion_radius)
x_grid = torch.logspace(-3, 3, 1000)
y_grid = coulomb_exclude.lr_from_dist(x_grid) + coulomb_exclude.sr_from_dist(x_grid)
# create a spline potential for with the exclusion range built in
spline = SplinePotential(
r_grid=x_grid, y_grid=y_grid, smearing=smearing, reciprocal=True, yhat_at_zero=0.0
)
t_grid = torch.logspace(-3, 3, 1000)
y_bare = coulomb.lr_from_dist(t_grid)
y_spline = spline.lr_from_dist(t_grid)
fig, ax = plt.subplots(
1, 1, figsize=(4, 3), sharey=True, sharex=True, constrained_layout=True
)
ax.plot(t_grid, y_spline, "b-", label=f"{exclusion_radius}Å exclusion (spline)")
ax.plot(t_grid, y_bare, "k:", label="smooth Coulomb")
ax.set_xlabel(r"$r$ / Å")
ax.set_ylabel(r"$V$ / a.u.")
ax.set_xlim(0, 20)
ax.set_ylim(0, 1.75)
ax.legend()
fig.show()
Compute the potential on a mesh¶
We use MeshInterpolator
and KSpaceFilter
to compute the potential on a grid.
# Determines grid resolution and initialize utility classes
ns = torchpme.lib.kvectors.get_ns_mesh(cell, smearing * 0.5)
MI = torchpme.lib.MeshInterpolator(
cell=cell, ns_mesh=ns, interpolation_nodes=3, method="P3M"
)
KF = torchpme.lib.KSpaceFilter(
cell=cell,
ns_mesh=ns,
kernel=spline,
fft_norm="backward",
ifft_norm="forward",
)
# Computes particle density on the grid (weighted by the "charges")
MI.compute_weights(positions)
rho_mesh = MI.points_to_mesh(particle_weights=charges)
# Computes the potential using the Fourier filter
ivolume = torch.abs(cell.det()).pow(-1)
potential_mesh = KF(rho_mesh) * ivolume
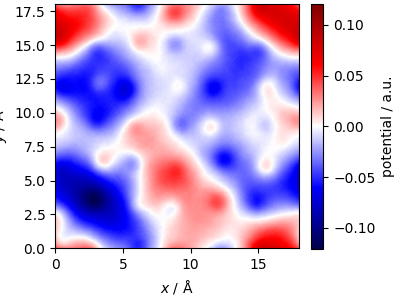
Plotting a slice of the potential demonstrates the smoothness of the potential, as the “core” region is damped out.
fig, ax = plt.subplots(
1, 1, figsize=(4, 3), sharey=True, sharex=True, constrained_layout=True
)
mesh_extent = [
0,
cell[0, 0],
0,
cell[1, 1],
]
z_plot = potential_mesh[0, :, :, 0].cpu().detach().numpy()
z_plot = np.vstack([z_plot, z_plot[0, :]]) # Add first row at the bottom
z_plot = np.hstack(
[z_plot, z_plot[:, 0].reshape(-1, 1)]
) # Add first column at the right
z_min, z_max = (z_plot.min(), z_plot.max())
z_range = max(abs(z_min), abs(z_max))
cf = ax.imshow(
z_plot,
extent=mesh_extent,
vmin=-z_range,
vmax=z_range,
origin="lower",
interpolation="bilinear",
cmap="seismic",
)
ax.set_xlabel(r"$x$ / Å")
ax.set_ylabel(r"$y$ / Å")
fig.colorbar(cf, label=r"potential / a.u.")
fig.show()
Atom-centered grids¶
To evaluate LODE features, we have to now project the potential within an atom-centered region. To do this, we define an atom-centered grid. Note that the quadrature here is not especially smart, and is only used for demonstrative purposes.
def get_theta_phi_quadrature(L):
"""Legendre quadrature nodes for integrals over theta, phi"""
quads = []
weights = []
for w_index in range(0, 2 * L - 1):
w = 2 * np.pi * w_index / (2 * L - 1)
roots_legendre_now, weights_now = scipy.special.roots_legendre(L)
all_v = np.arccos(roots_legendre_now)
for v, weight in zip(all_v, weights_now, strict=True):
quads.append([v, w])
weights.append(weight)
norm = 4 * torch.pi / np.sum(weights)
return torch.tensor(quads), torch.tensor(weights) * norm
def get_radial_quadrature(order, R):
"""
Generates Gauss-Legendre quadrature nodes and weights for radial integration
in spherical coordinates over the interval [0, R].
"""
gl_nodes, gl_weights = np.polynomial.legendre.leggauss(order)
nodes = (R / 2) * (gl_nodes + 1)
weights = (R / 2) ** 3 * gl_weights * (gl_nodes + 1) ** 2
return torch.from_numpy(nodes), torch.from_numpy(weights)
def get_full_grid(n, R):
lm_nodes, lm_weights = get_theta_phi_quadrature(n)
r_nodes, r_weights = get_radial_quadrature(n, R)
full_weights = (r_weights.reshape(-1, 1) * lm_weights.reshape(1, -1)).flatten()
cos_nodes = torch.cos(lm_nodes[:, 0]).reshape(1, -1)
sin_nodes = torch.sin(lm_nodes[:, 0]).reshape(1, -1)
xyz_nodes = torch.vstack(
[
(r_nodes.reshape(-1, 1) * cos_nodes).flatten(),
(
r_nodes.reshape(-1, 1) * (sin_nodes * torch.cos(lm_nodes[:, 1]))
).flatten(),
(
r_nodes.reshape(-1, 1) * (sin_nodes * torch.sin(lm_nodes[:, 1]))
).flatten(),
]
).T
return xyz_nodes, full_weights
xyz, weights = get_full_grid(3, exclusion_radius / 4)
The grid can then be centered on each atom, and the
back-interpolation of MeshInterpolator be used to
evaluate the potential values
The grid can be shown in the context of the atomic structure
dummy = ase.Atoms(positions=grid_i.numpy(), symbols="H" * len(grid_i))
chemiscope.show(
frames=[structure + dummy],
properties={
"potential": {
"target": "atom",
"values": np.concatenate([[0] * len(positions), pots_i.flatten().numpy()]),
},
"grid weights": {
"target": "atom",
"values": np.concatenate([[0] * len(positions), weights.flatten().numpy()]),
},
},
mode="structure",
settings=chemiscope.quick_settings(
structure_settings={
"unitCell": True,
"bonds": False,
"environments": {"activated": False},
"color": {
"property": "potential",
"min": -0.15,
"max": 0.15,
"transform": "linear",
"palette": "seismic",
},
}
),
environments=chemiscope.all_atomic_environments([structure + dummy]),
)

/home/runner/work/torch-pme/torch-pme/examples/07-lode-demo.py:303: UserWarning: `frames` argument is deprecated, use `structures` instead
chemiscope.show(
Computing the projection¶
In order to compute the LODE coefficients, we simply have to evaluate the basis on the same atom-centered grid. Here for example we just use \((1,x,y,z)\) as basis
# define the basis
f0 = torch.ones(len(xyz))
fx = xyz[:, 0]
fy = xyz[:, 1]
fz = xyz[:, 2]
# normalize the basis
f0 = f0 / torch.sqrt((weights * f0**2).sum())
fx = fx / torch.sqrt((weights * fx**2).sum())
fy = fy / torch.sqrt((weights * fy**2).sum())
fz = fz / torch.sqrt((weights * fz**2).sum())
# compute
lode_i = torch.tensor(
[
(weights * f0 * pots_i).sum(),
(weights * fx * pots_i).sum(),
(weights * fy * pots_i).sum(),
(weights * fz * pots_i).sum(),
]
).squeeze()
print(f"LODE features: {lode_i}")
LODE features: tensor([-0.4658, -0.0362, 0.0017, 0.0364], dtype=torch.float64)
Defines a LODE calculator¶
All these pieces can be combined in a relatively concise Calculator
class that computes LODE features.
class SmoothCutoffCoulomb(SplinePotential):
def __init__(
self, smearing: float, exclusion_radius: float, n_points: int | None = 1000
):
coulomb = CoulombPotential(smearing=smearing, exclusion_radius=exclusion_radius)
x_grid = torch.logspace(-3, 3, n_points)
y_grid = coulomb.lr_from_dist(x_grid) + coulomb.sr_from_dist(x_grid)
super().__init__(
r_grid=x_grid,
y_grid=y_grid,
smearing=smearing,
exclusion_radius=exclusion_radius,
reciprocal=True,
yhat_at_zero=0.0,
)
class LODECalculator(torchpme.Calculator):
"""
Compute expansions of the local potential in an atom-centered basis.
:param potential: A :class:`Potential` implementing the convolution
kernel. Real-space components are not used.
:param n_grid: Atom-centered grid size; this is the number of nodes per
dimension, so the overall number of points is ``n_grid**3``.
"""
def __init__(self, potential: Potential, n_grid: int = 3):
super().__init__(potential=potential)
assert self.potential.exclusion_radius is not None
assert self.potential.smearing is not None
cell = torch.eye(3)
ns = torch.tensor([2, 2, 2])
self._MI = torchpme.lib.MeshInterpolator(
cell=cell, ns_mesh=ns, interpolation_nodes=3, method="P3M"
)
self._KF = torchpme.lib.KSpaceFilter(
cell=cell,
ns_mesh=ns,
kernel=self.potential,
fft_norm="backward",
ifft_norm="forward",
)
# assumes a smooth exclusion region so sets the integration cutoff to half that
nodes, weights = get_full_grid(n_grid, potential.exclusion_radius / 2)
# these are the "stencils" used to project the potential
# on an atom-centered basis. NB: weights might also be incorporated
# in here saving multiplications later on
stencils = [
(nodes[:, 0] * 0.0 + 1.0) / torch.sqrt((weights).sum()), # constant
(nodes[:, 0]) / torch.sqrt((weights * nodes[:, 0] ** 2).sum()), # x
(nodes[:, 1]) / torch.sqrt((weights * nodes[:, 1] ** 2).sum()), # y
(nodes[:, 2]) / torch.sqrt((weights * nodes[:, 2] ** 2).sum()), # z
]
self._basis = torch.stack(stencils)
self._nodes = nodes
self._weights = weights
def forward(
self,
charges: torch.Tensor,
cell: torch.Tensor,
positions: torch.Tensor,
neighbor_indices: torch.Tensor | None = None,
neighbor_distances: torch.Tensor | None = None,
periodic: torch.Tensor | None = None,
node_mask: torch.Tensor | None = None,
pair_mask: torch.Tensor | None = None,
kvectors: torch.Tensor | None = None,
) -> torch.Tensor:
# Update meshes
assert self.potential.smearing is not None # otherwise mypy complains
ns = torchpme.lib.kvectors.get_ns_mesh(cell, self.potential.smearing / 2)
self._MI.update(cell, ns)
self._KF.update(cell, ns)
# Compute potential
self._MI.compute_weights(positions)
rho_mesh = self._MI.points_to_mesh(particle_weights=charges)
ivolume = torch.abs(cell.det()).pow(-1)
potential_mesh = self._KF(rho_mesh) * ivolume
# Places integration grids around each atom
all_points = torch.stack([self._nodes + pos for pos in positions]).reshape(
-1, 3
)
# Evaluate the potential on the grids
self._MI.compute_weights(all_points)
all_potentials = self._MI.mesh_to_points(potential_mesh).reshape(
len(positions), len(self._nodes), -1
)
# Compute lode as an integral
return torch.einsum("ijq,bj,j->ibq", all_potentials, self._basis, self._weights)
Instantiates the calculator and evaluates it for the NaCl structure
smearing = 0.5
exclusion_radius = 8.0
my_pot = SmoothCutoffCoulomb(smearing=smearing, exclusion_radius=exclusion_radius)
my_lode = LODECalculator(potential=my_pot, n_grid=8)
lode_features = my_lode.forward(
charges=charges, cell=cell, positions=positions
).squeeze()
The basis function hardcoded in the LODECalculator class have a scalar (mean potential) and vectorial (roughly corresponding to the mean electric field) nature, so we can plot it with color corresponding to the constant part, and arrows proportional to the vectorial component.
def value_to_seismic(value, vrange=0.1):
"""Map values to RGB color string using the 'seismic' colormap."""
vmin, vmax = -vrange, vrange
# Ensure the value is within the specified range
clipped_value = np.clip(value, vmin, vmax)
norm = (clipped_value - vmin) / (vmax - vmin)
rgba = matplotlib.colormaps["seismic"](norm)
rgb = tuple(int(255 * c) for c in rgba[:3])
return "#{:02x}{:02x}{:02x}".format(*rgb)
chemiscope.show(
frames=[structure],
properties={
"lode[1]": {
"target": "atom",
"values": np.concatenate([lode_features[:, 0].flatten().numpy()]),
},
"lode[x]": {
"target": "atom",
"values": np.concatenate([lode_features[:, 1].flatten().numpy()]),
},
"lode[y]": {
"target": "atom",
"values": np.concatenate([lode_features[:, 2].flatten().numpy()]),
},
"lode[z]": {
"target": "atom",
"values": np.concatenate([lode_features[:, 3].flatten().numpy()]),
},
},
shapes={
"lode": {
"kind": "arrow",
"parameters": {
"global": {
"baseRadius": 0.2,
"headRadius": 0.3,
"headLength": 0.5,
},
"atom": [
{
"vector": (4 * lode_features[i, 1:]).tolist(),
"color": value_to_seismic(lode_features[i, 0], 0.6),
}
for i in range(len(lode_features))
],
},
}
},
mode="structure",
settings=chemiscope.quick_settings(
structure_settings={
"unitCell": True,
"bonds": False,
"environments": {"activated": False},
"shape": ["lode"],
}
),
environments=chemiscope.all_atomic_environments([structure]),
)
/home/runner/work/torch-pme/torch-pme/examples/07-lode-demo.py:507: UserWarning: `frames` argument is deprecated, use `structures` instead
chemiscope.show(
Total running time of the script: (0 minutes 2.780 seconds)