Note
Go to the end to download the full example code.
Optimizing a linear combination of potentials¶
- Authors:
Egor Rumiantsev @E-Rum; Philip Loche @PicoCentauri
This is an example to demonstrate the usage of the CombinedPotential class to
evaluate potentials that combine multiple pair potentials with optimizable weights.
We will optimize the weights to reporoduce the energy of a system that
interacts solely via Coulomb interactions.
import ase.io
import chemiscope
import matplotlib.pyplot as plt
import torch
from vesin.torch import NeighborList
from torchpme import CombinedPotential, EwaldCalculator, InversePowerLawPotential
from torchpme.prefactors import eV_A
dtype = torch.float64
Combined potentials¶
We load the small dataset that contains eight
randomly placed point charges in a cubic cell of different cell sizes. Each structure
contains four positive and four negative charges that interact via a Coulomb
potential.
frames = ase.io.read("coulomb_test_frames.xyz", ":")
chemiscope.show(
frames=frames,
mode="structure",
settings=chemiscope.quick_settings(
structure_settings={"unitCell": True, "bonds": False}
),
)

/home/runner/work/torch-pme/torch-pme/examples/08-combined-potential.py:44: UserWarning: `frames` argument is deprecated, use `structures` instead
chemiscope.show(
We choose half of the box length as the cutoff for the neighborlist and also
deduce the other parameters from the first frame.
We now construct the potential as sum of two InversePowerLawPotential using
CombinedPotential. The presence of a numerical smearing value is used as an
indication that the potential can compute the terms needed for range-separated
evaluation, and so one has to set it also for the combined potential, even if it is
not used explicitly in the evaluation of the combination.
pot_1 = InversePowerLawPotential(exponent=1, smearing=smearing, prefactor=eV_A)
pot_2 = InversePowerLawPotential(exponent=2, smearing=smearing, prefactor=eV_A)
pot_1 = pot_1.to(dtype=dtype)
pot_2 = pot_2.to(dtype=dtype)
potential = CombinedPotential(potentials=[pot_1, pot_2], smearing=smearing)
potential = potential.to(dtype=dtype)
# Note also that :class:`CombinedPotential` can be used with any combination of
# potentials, as long they are all either direct or range separated. For instance, one
# can combine a :class:`CoulombPotential` and a :class:`SplinePotential`.
Plotting terms in the potential¶
We now plot of the individual and combined potential functions together with an
explicit sum of the two potentials.
dist = torch.logspace(-3, 2, 1000, dtype=dtype)
fig, ax = plt.subplots()
ax.plot(dist, pot_1.from_dist(dist), label="p=1")
ax.plot(dist, pot_2.from_dist(dist), label="p=2")
ax.plot(dist, potential.from_dist(dist).detach(), label="Combined potential", c="black")
ax.plot(
dist,
pot_1.from_dist(dist) + pot_2.from_dist(dist),
label="Explict combination",
ls=":",
)
ax.set(
xlabel="Distance", ylabel="Potential", xscale="log", yscale="log", xlim=[1e-3, 1e2]
)
ax.legend()
plt.show()
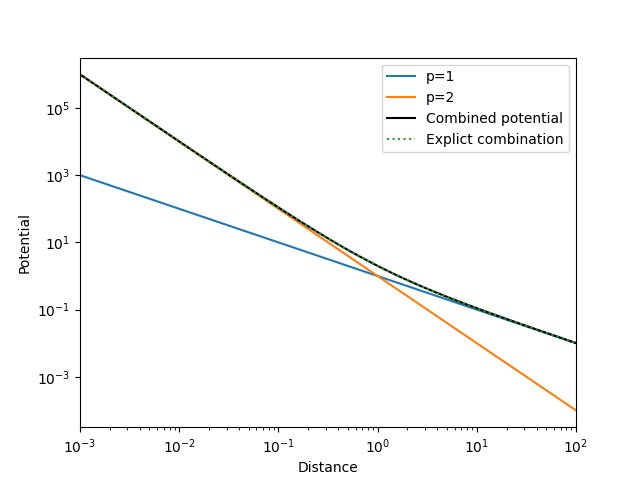
In the log-log plot we see that the \(p=2\) potential (orange) decays much faster compared to the \(p=1\) potential (blue). We also verify that the combined potential (black) is the sum of the two potentials that we explicitly calculated (dotted green line).
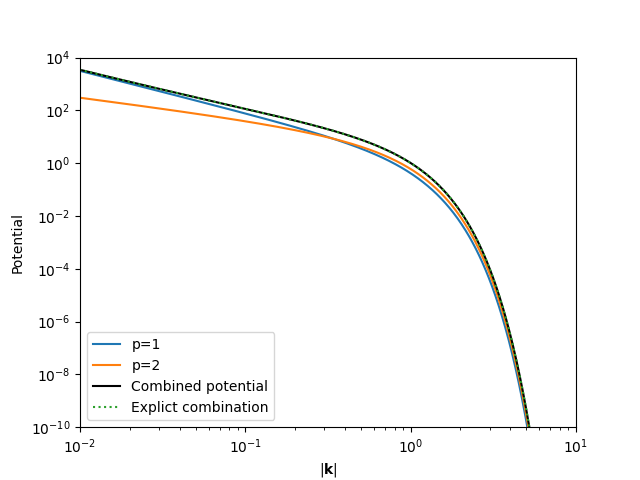
The CombinedPotential class
combines all terms in a range-separated potential, including the k-space
kernel.
k = torch.logspace(-2, 2, 1000, dtype=dtype)
fig, ax = plt.subplots()
ax.plot(dist, pot_1.lr_from_k_sq(k**2), label="p=1")
ax.plot(dist, pot_2.lr_from_k_sq(k**2), label="p=2")
ax.plot(
dist, potential.lr_from_k_sq(k**2).detach(), label="Combined potential", c="black"
)
ax.plot(
dist,
pot_1.lr_from_k_sq(k**2) + pot_2.lr_from_k_sq(k**2),
label="Explict combination",
ls=":",
)
ax.set(
xlabel=r"$|\mathbf{k}|$",
ylabel="Potential",
xscale="log",
yscale="log",
xlim=[1e-2, 1e1],
ylim=[1e-10, 1e4],
)
ax.legend()
plt.show()
Optimizing the mixing weights¶
We next construct the calculator. Note that below we use the EwaldCalculator
but one can of course also use the PMECalculator if one wants to optimize a
much bigger system.
calculator = EwaldCalculator(potential=potential, lr_wavelength=lr_wavelength)
calculator.to(dtype=dtype)
EwaldCalculator(
(potential): CombinedPotential(
(potentials): ModuleList(
(0-1): 2 x InversePowerLawPotential()
)
)
)
To save some time during optimization we precompute the neighborlist and store all values in convient lists. We store the data in lists of torch tensors because in general the number of particles in each frame can be different.
nl = NeighborList(cutoff=cutoff, full_list=False)
l_positions = []
l_cell = []
l_charges = []
l_neighbor_indices = []
l_neighbor_distances = []
l_ref_energy = torch.zeros(len(frames))
for i_atoms, atoms in enumerate(frames):
positions = torch.from_numpy(atoms.positions)
cell = torch.from_numpy(atoms.cell.array)
charges = torch.from_numpy(atoms.get_initial_charges()).reshape(-1, 1)
p, d = nl.compute(points=positions, box=cell, periodic=True, quantities="Pd")
l_positions.append(positions)
l_cell.append(cell)
l_charges.append(charges)
l_neighbor_indices.append(p)
l_neighbor_distances.append(d)
l_ref_energy[i_atoms] = atoms.get_potential_energy()
Definition of loss and optimizer¶
For the optimization we define two functions that compute the energy of all structures and the mean squared error of the energy with respect to the reference values as loss.
def compute_energy() -> torch.Tensor:
"""Compute the energy of all structures using a globally defined `calculator`."""
energy = torch.zeros(len(frames))
for i_atoms in range(len(frames)):
charges = l_charges[i_atoms]
potential = calculator(
charges=charges,
cell=l_cell[i_atoms],
positions=l_positions[i_atoms],
neighbor_indices=l_neighbor_indices[i_atoms],
neighbor_distances=l_neighbor_distances[i_atoms],
)
energy[i_atoms] = (charges * potential).sum()
return energy
def loss() -> torch.Tensor:
"""Compute the mean squared error of the energy."""
energy = compute_energy()
mse = torch.sum((energy - l_ref_energy) ** 2)
return mse.sum()
optimizer = torch.optim.Adam(calculator.parameters(), lr=0.1)
Running the optimization¶
We now optimize the weights of the potentials to minimize the mean squared error using
the torch.optim.Adam optimizer and stop either after 1000 epochs or when the
loss is smaller than \(10^{-2}\).
weights_timeseries = []
loss_timeseries = []
for _ in range(1000):
optimizer.zero_grad()
loss_value = loss()
loss_value.backward()
optimizer.step()
loss_timeseries.append(float(loss_value.detach().cpu()))
weights_timeseries.append(calculator.potential.weights.detach().cpu().tolist())
if loss_value < 1e-4:
break
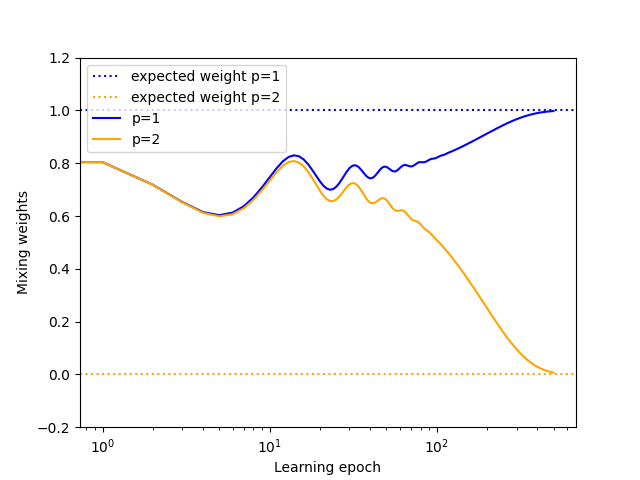
We can show the evolution of the weights during the optimization. The weights for the \(1/r\) and \(1/r^2\) potentials converge towards \(1\) and \(0\), respectively. This is the expected behavior, since the reference potential used to compute the energy of the structures includes only a Coulombic term.
fig, ax = plt.subplots()
ax.axhline(1, c="blue", ls="dotted", label="expected weight p=1")
ax.axhline(0, c="orange", ls="dotted", label="expected weight p=2")
weights_timeseries_array = torch.tensor(weights_timeseries)
ax.plot(weights_timeseries_array[:, 0], label="p=1", c="blue")
ax.plot(weights_timeseries_array[:, 1], label="p=2", c="orange")
ax.set(
ylim=(-0.2, 1.2),
xlabel="Learning epoch",
ylabel="Mixing weights",
xscale="log",
)
ax.legend()
plt.show()
Total running time of the script: (0 minutes 51.916 seconds)